
From platform to product: Accelerating time-to-market for platform technologies
Featured in ONdrugDelivery, Fran Pencliffe explores the benefits of platform devices for parenteral delivery and outlines the challenges, risks and best practices when bringing a combination product to market in this way.
Platform devices have long been considered the “holy grail” of drug delivery device design. The appeal of platforms is clear, with companies looking to create innovative platforms to meet the evolving requirements of new therapies, while pharma companies are looking to use these technologies to expedite combination product development.
Defining platform devices in drug delivery
In the drug delivery industry, the term “platform devices” encompasses off-the-shelf prefilled syringes, fixed- or variable dose pen injectors, autoinjectors for “standard” volumes of “low”-viscosity formulations and higher-volume on-body delivery systems. Platforms are also being developed to handle high-viscosity formulations or support automatic drug reconstitution, making technology selection increasingly complex.
“The core feature of a platform is a consistent device architecture, with customisation options to accommodate VARYING assets, user groups or branding.”
Unlike devices developed for a single formulation, platforms are designed for use with multiple drug assets with varying requirements, such as different dose volumes, viscosities, user groups and use environments (Figure 1). The core feature of a platform is a consistent device architecture, with customisation options to accommodate varying assets, user groups or branding. Platforms vary from “narrow” (devices catering to very similar drug profiles) to “broad” (those intended for diverse therapy areas, user groups and drug properties). Broader platforms, while targeting a larger market, present greater technical challenges and risks during both platform and combination product development.
When designed and implemented correctly, platform devices offer numerous benefits for both device developers and pharmaceutical companies.
The benefits and risks of platform devices
For those designing a platform device, the benefits are clear. A common architecture can be used with multiple drug products, increasing the potential market size for a single development effort. This reduces the investment cost per marketed drug and simplifies the process of navigating the intellectual property landscape for each new asset. Additionally, economies of scale in manufacturing components lower the cost per device, making the device more attractive to potential partners. However, high rewards often come with high risk, depending on the targeted platform.
Proper development and characterisation of a platform technology often requires significant upfront investment from the device developer, which may be made at risk prior to establishing a partnership with a pharmaceutical company. This can be challenging and relies on an “if you build it, they will come” mentality, often involving millions of dollars with no guaranteed return.
For pharmaceutical companies, platform devices offer a near “off-the-shelf” solution to deliver their assets. Using an existing (and hopefully already marketed) device can minimise time-to-market and the risks associated with developing a new device by building the combination product on proven technology. However, selecting the wrong device can lead to extensive device modifications or starting over with a new device, both of which may extend the development timeline and delay product launch. There are, however, ways to mitigate these risks and realise the benefits of platform devices.
Key strategies for successful platform development
To maximise return on investment when designing a platform technology, there are two key recommendations: understanding the target market to define an achievable platform boundary and preparing a data pack to minimise the effort required for potential partners to use the device.
The first challenge in platform device development is often generating the necessary investment required. To demonstrate a potential return on investment, it is critical to research upcoming drug pipelines and identify groups of assets that are likely to have similar delivery requirements. This can be done by examining Phase I and II trial data and monitoring trends in growing therapy areas. A broad potential portfolio strengthens the case for creating a platform design and maximises the likelihood of securing development investment.
“A platform with a broad performance envelope is likely to have the largest market potential but will be riskier and costlier to develop.”
Once this target drug portfolio is identified, use the likely delivery requirements to define the platform’s boundaries. For example, consider whether the target therapies are intended for intramuscular or subcutaneous delivery, the expected volumes and viscosities that the platform will need to accommodate, and whether a fixed or user-selectable dose is needed. A platform with a broad performance envelope is likely to have the largest market potential but will be riskier and costlier to develop. A device concept is unlikely to gain significant attention from potential partners until functional performance can be readily proven, so clearly defining the platform performance envelope early and sticking to it throughout development will be the fastest route to market.
When developing a platform, it is also recommended to develop a data pack for potential partners to review as part of a technical due diligence. Sharing test data is the most compelling argument when selling a technology. Demonstrating that the device can, for example, deliver the correct volume and viscosity in the correct time instils confidence in its performance, which cannot be replicated through modelling or simulation. Although this requires effort in prototyping and developing test methods, the increase in “selling power” from having this real-world data increases the likelihood of a return on investment.
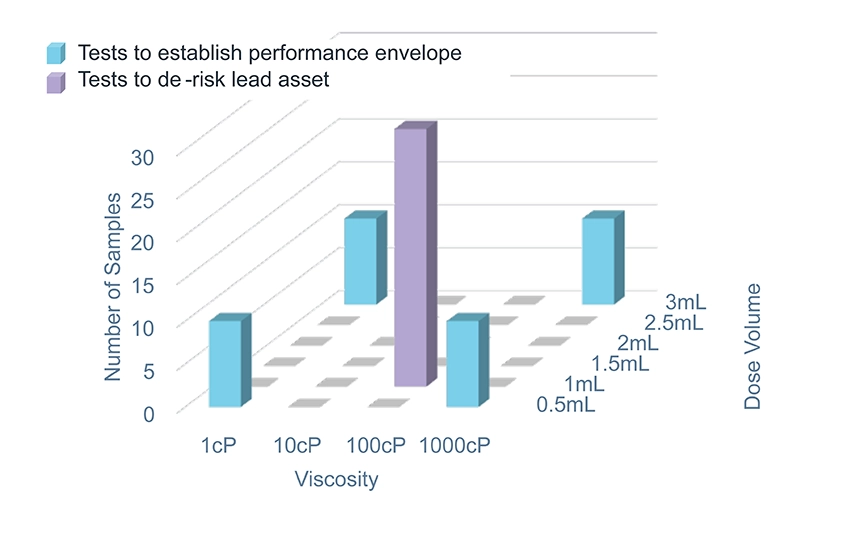
For a platform product, it is good practice to create a platform test plan with low-fidelity testing at the edges of the performance range to give confidence in the platform boundaries and high-fidelity (verification) testing on one or two specific configurations that represent the most likely assets in the target pipeline. Figure 2 shows an example of how the fidelity of testing can be adjusted to provide confidence in the platform envelope while focusing effort on the lead asset. Offering potential partners the opportunity to test their formulation in the device, with sample devices available for filling and existing test methods, allows for quick and cost-effective testing.
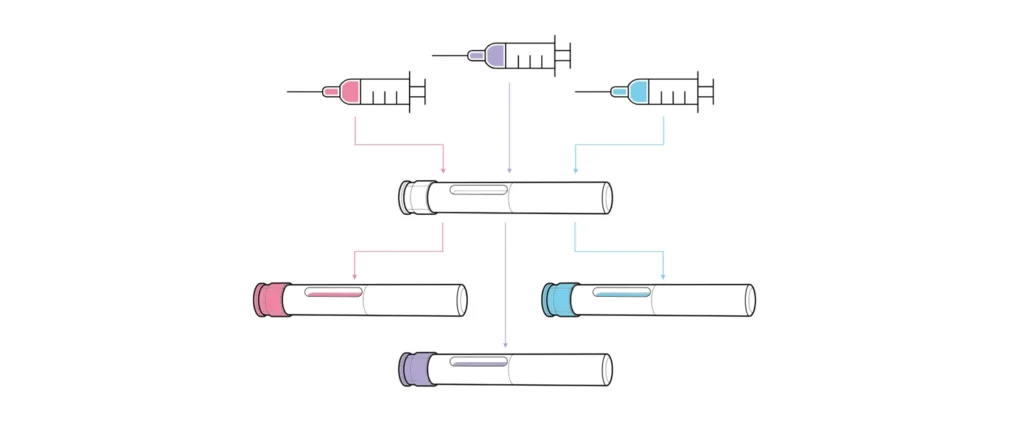
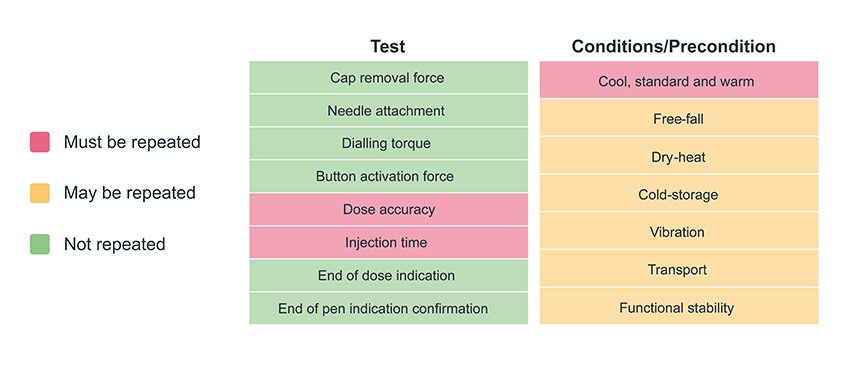
Of course, there is no such thing as a truly “off-the-shelf” platform product, so the second critical aspect of the data pack to share with potential partners is the bridging plan. Minimising and clearly defining the design work and associated testing to be repeated for each new asset reduces time-to-market and further increases confidence in the device developer’s ability to deliver on a combination product development programme. Figure 3 shows an example of a bridging test plan to convert from a platform injection device to a combination product – note that the specifics will be highly dependent on the drug and device in question.
By understanding the target market and device boundaries and creating a data pack to convey the platform’s benefits to potential partners, the potential market size for a platform can be maximised and the potential return on the initial development effort increased.
Choosing the right platform for the target drug pipeline
For pharmaceutical companies seeking a platform device to fit the delivery requirements of as many assets as possible in a drug pipeline, the critical activities are understanding the formulations, the available and applicable technologies and using existing data to minimise time-to-market.
“Before searching for a device technology, it is vital to understand the requirements of the target drug assets.”
Before searching for a device technology, it is vital to understand the requirements of the target drug assets. Pharmaceutical companies should identify groups of assets with similar characteristics and intended use profiles across their portfolios, for example, all those intended for subcutaneous injection in a home environment. This enables them to search for platforms with the correct performance envelope, assessing technologies not just for the lead asset but with the wider portfolio in mind, thereby offering the potential to minimise time-to-market for future assets.
It is also crucial to understand what the drugs require from a device as much as possible. What is the dose volume? What is the formulation viscosity, and how does it change with temperature and shear rate? What is the target delivery time? Answering as many questions about the required performance of a platform as early as possible can help optimise the search process and enable the device developer to gather and present the most relevant data during the due diligence process.
Another important process for pharmaceutical companies to undertake is to survey the technology landscape by searching for existing devices that meet the formulation’s needs. This creates a shortlist of devices to be investigated further through supplier contact and deeper dives into the device data package. The primary focus during this survey is to establish device compatibility with the lead asset, with a secondary focus on compatibility with the wider drug wider pipeline.
To gain confidence in a device’s ability to support the lead asset, pharmaceutical companies should look for empirical evidence wherever possible. Clear usability and test data supported by robust test methodology is the strongest indicator of device performance, while tolerance analyses and mathematical models can evidence a device’s ability to perform at scale. Ideally, the test data should showcase a device’s ability to deliver a formulation similar to the lead asset across all appropriate preconditions, for example, free-fall is often a point of failure for injection devices, or else provide explanations for any expected risks and mitigations.
The next step is to review the manufacturing and assembly plan to ensure that device supply can scale reliably and securely to meet expected market volumes at the required price point. Where possible, all evidence in the design history file should be reviewed for direct applicability to the asset under development, such as which test results can be used as part of a combination product submission, which need to be repeated and how well defined the scope of any work that needs to be repeated is.
“A strong device partner will demonstrate a clear and in-depth understanding of their platform and technology, with readily available evidence or a plan to gather this evidence and the expected risks.”
To assess the platform as a whole, pharmaceutical companies should focus on the boundaries of performance, such as range of volumes and viscosities supported, and how well the device developer understands these boundaries. Can both the maximum volume and viscosity be delivered in the required time by a single device under all conditions? What evidence supports this? What parts need to be changed to support different configurations, and how much investment is needed to meet those requirements within the desired timeline? A strong device partner will demonstrate a clear and in-depth understanding of their platform and technology, with readily available evidence or a plan to gather this evidence and the expected risks. Replacing test data with simulation data is adequate for early stage devices but does not fully mitigate the risk of a device underperforming and requiring more development work. If test data is not provided or fully documented, it indicates that the device is early in the development process and not “ready to use”. Any first-time tests are likely to show failures and trigger a design loop. If this testing has not been conducted properly, extensive development work is likely still required within the platform development, posing a risk to time-to-market and increasing costs.
Integrating device and drug: steps to market readiness
Once compatibility between a device and a drug has been established, a risk assessment should be conducted as part of the creation of a plan for customising and verifying the combination product. Existing test results can be used if there is sufficient evidence that the drug will not influence the outcomes, such as cap removal force if the same components are being used, or free-fall preconditioning if the drug density matches that used in testing. The tests that are likely to need to be repeated in all cases include dose accuracy under standard, warm and cool preconditions (Figure 3). However, methods, fixtures and processes can be reused if dose accuracy testing has been conducted previously. This process allows for the minimum viable test plan, drastically reducing the time and effort required to verify combination product performance compared with a custom development.
As platform devices are required to meet an ever-widening set of market demands, there is an increasing need to simplify the process of developing these devices and adopting them for combination products. Through independent characterisation of both device and drug, combination product development can be greatly simplified, reducing the time and investment required to bring a new therapy to market.
Connect with CDP
For more on how to accelerate time-to-market for platform drug delivery devices and combination products, contact Cambridge Design Partnership.