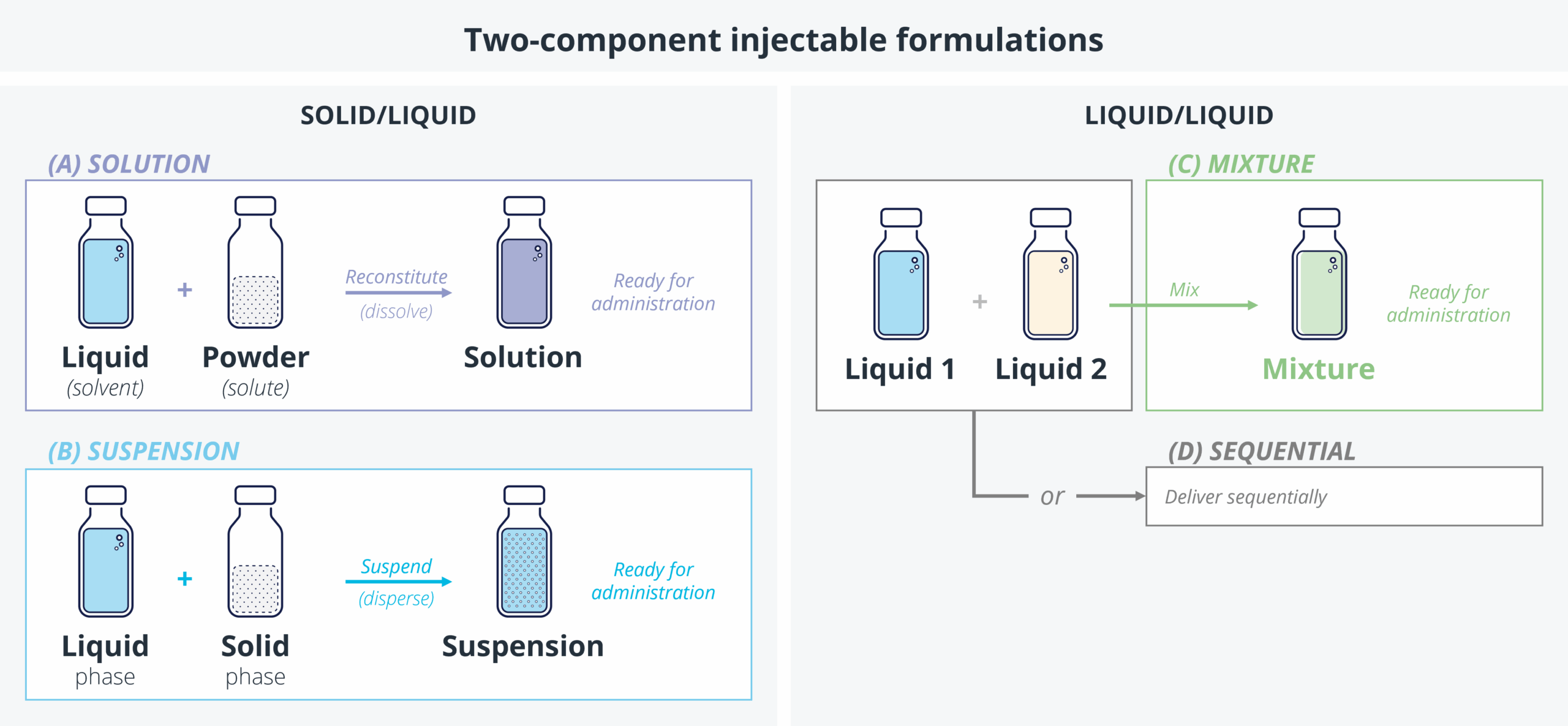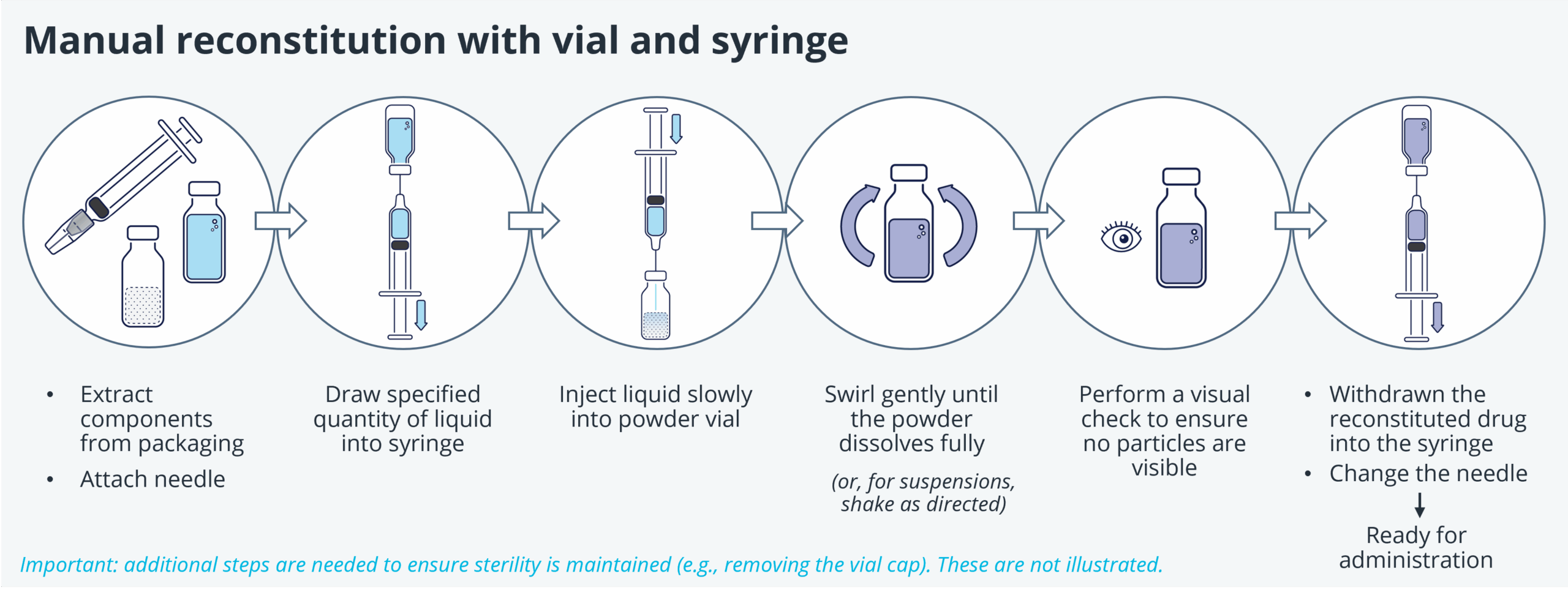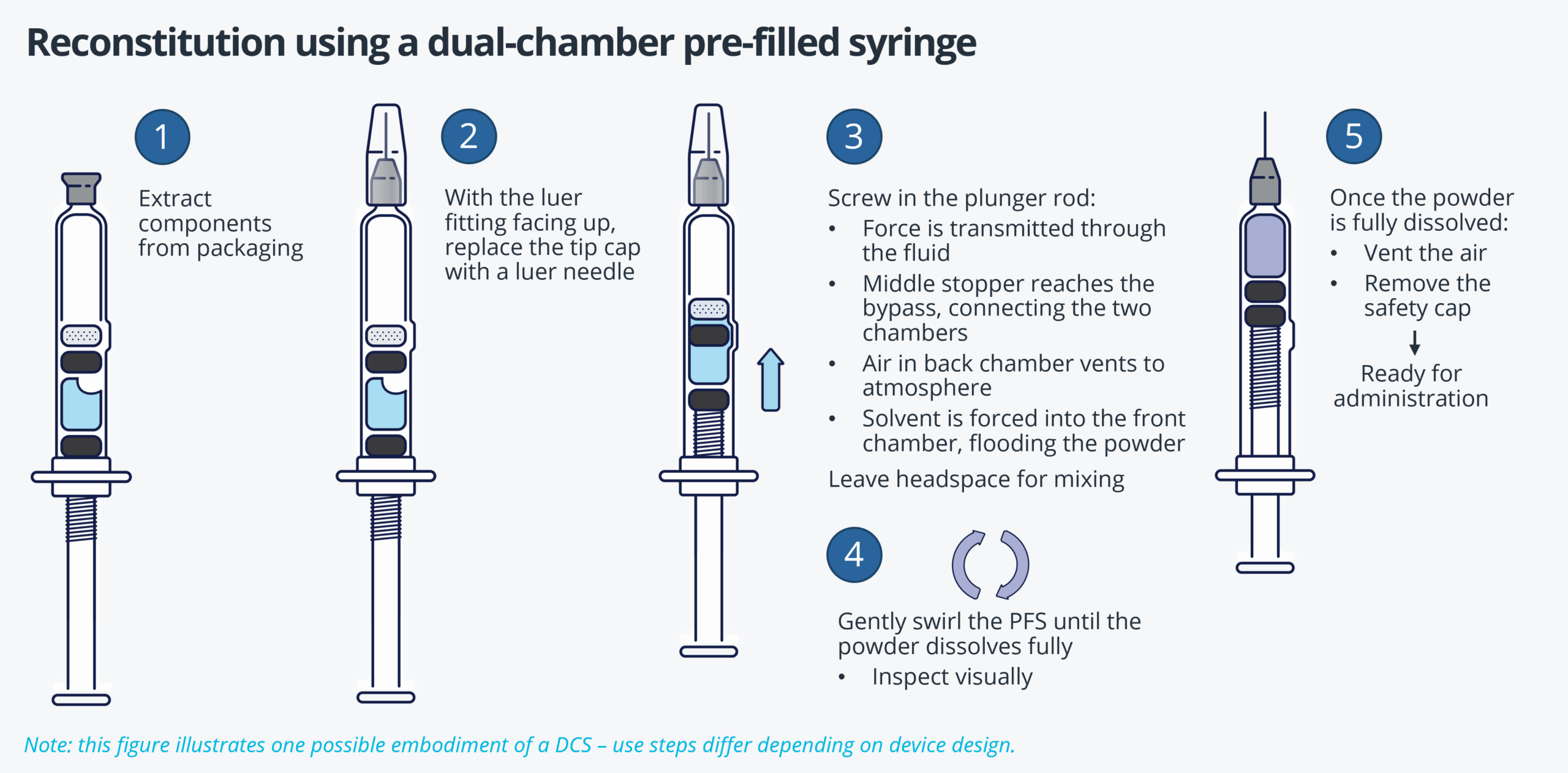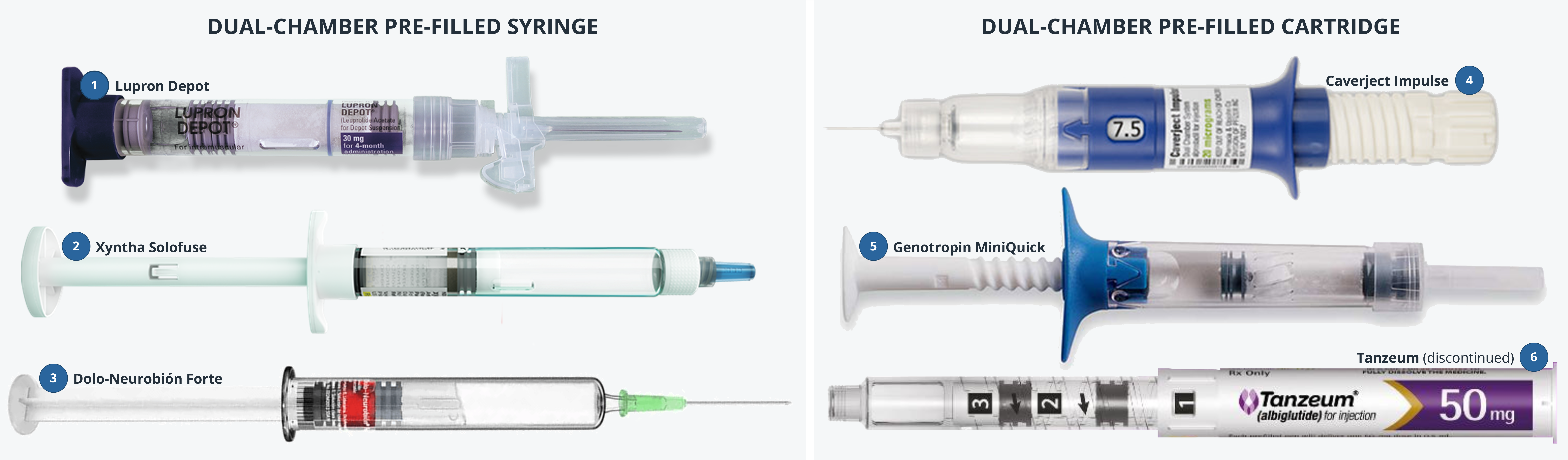


Introduction
Platform devices are often the preferred starting point for combination product development, and for good reason. A well-characterised platform offers a proven architecture, defined performance boundaries, and a clear path to market. However, an increasing number of therapies in development are pushing into territories where there is currently no existing platform to serve their needs.
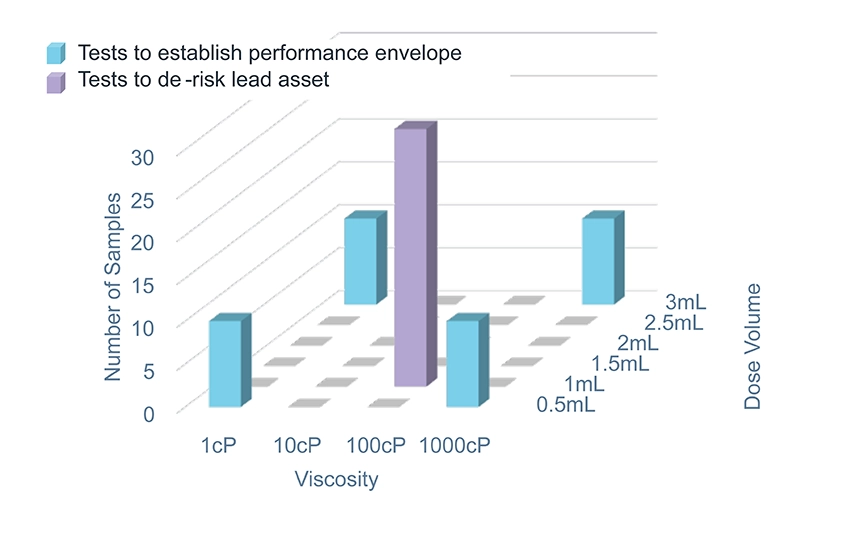
Novel biologics are extending the boundaries of volumes and viscosities, cell and gene therapies are leading to ever-more tissue-targeted deliveries, suspensions and co-delivered (or sequentially delivered) therapies are among those demanding entirely new thinking about how, where, and whether a drug can be delivered. These formulations can all be gathered under the umbrella term “specialty delivery” and are united by a common challenge – where there is no existing platform device able to deliver the formulation, a delivery system becomes a core part of formulation development and testing. Where traditional developments are often able to rely on vial-and-syringe delivery to enable early clinical data to be gathered, these specialty delivery developments (especially those requiring tissue-targeted delivery) require more sophisticated devices to be available earlier in the therapy or formulation’s life cycle.
For these developments, feasibility work looks very different to what we expect when adopting a platform. Rather than asking “Can this platform device deliver our formulation?”, the question becomes “Is it possible to deliver this formulation at all?”, which could mean the difference between a few weeks of benchtop testing and months or even years of exploratory research. Answering this new feasibility question efficiently requires a structured, pragmatic approach that balances scientific rigour with the realities of early-stage development timelines and budgets.
The framework described in this article has been developed through more than a decade of feasibility work on some of the most challenging drug delivery developments we’ve encountered; projects where the answer wasn’t obvious, and where finding it efficiently made the difference between a development moving forward and stalling.
This article explores how theoretical, synthetic and tissue models can be combined to de-risk novel drug delivery developments, and how to deploy them intelligently to turn an apparently impossible challenge into a feasible one. While the worked example we’ll draw on throughout – a novel device for drug delivery directly to the brain – sits at the more complex end of the spectrum, the principles apply equally to any development where an existing platform doesn’t fit, or where it isn’t yet clear whether a device is a good match for the therapy. The approach scales to the necessary level of complexity.
What Does Feasibility Really Mean?
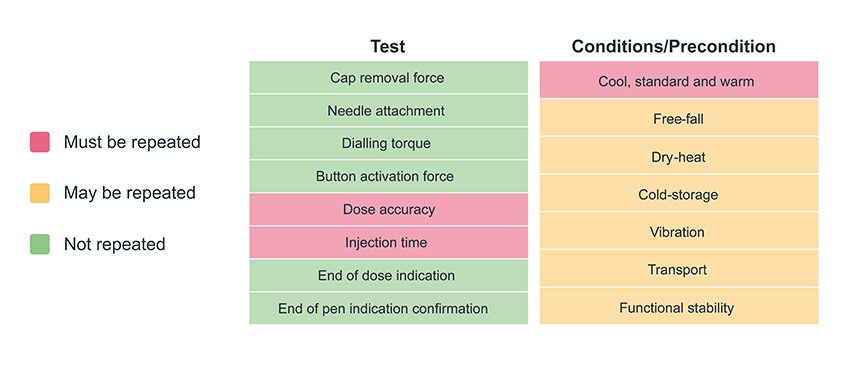
Feasibility for specialty delivery means different things depending on where you sit on the development complexity spectrum. At one end, it might mean confirming that an existing device can deliver a formulation that sits just outside its stated performance envelope, e.g., pushing the viscosity boundary and testing whether a longer injection time is still acceptable to users.
At the other end, feasibility might mean starting from scratch with a therapy that has never been delivered before, to a target site that has never been accessed in this way, using a device that doesn’t yet exist. Here, the risks are profound, and the unknowns are numerous, spanning therapy efficacy, navigation and targeting (including positional accuracy), tissue tolerability, usability, and many more.
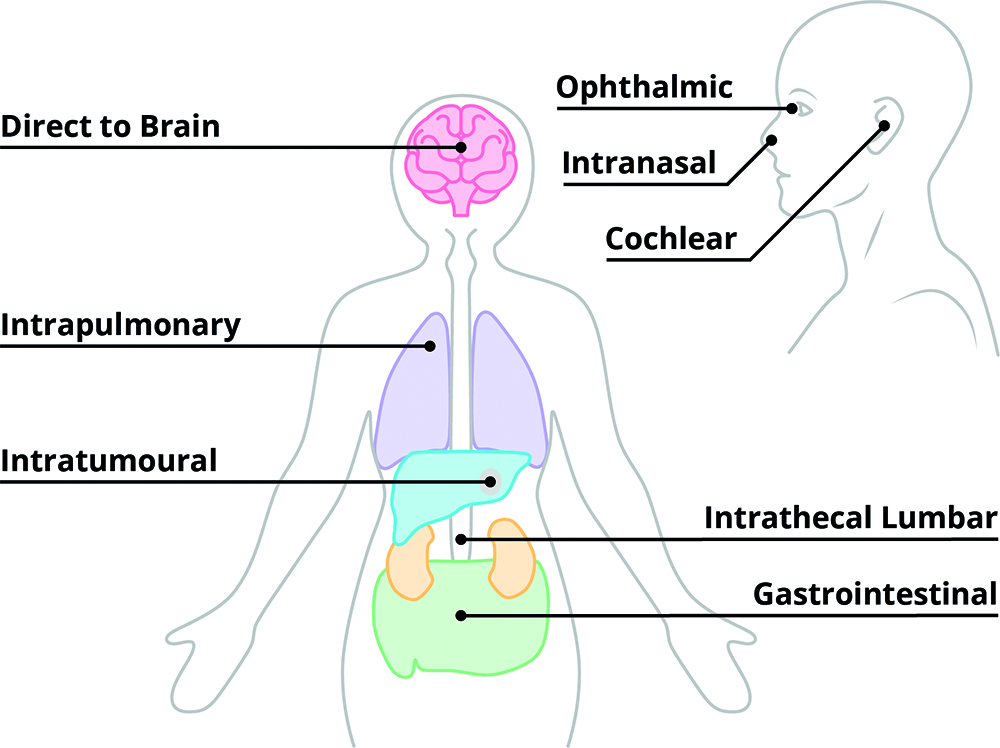
The first challenge is therefore to understand where your development sits on this spectrum, which can be done by identifying the key risks to be addressed. This starts by understanding what the needs of the formulation are as compared to existing formulations on the market or in clinic. If, for example, you are targeting subcutaneous delivery, chances are navigating the device to the right area is not going to be a huge risk. But if you’re targeting a specific structure of the brain or a nerve within the nasal cavity, suddenly how the formulation is delivered to the right location is an unknown factor that’s critical to the efficacy of the therapy. The same principles apply for other aspects – are you targeting a new user group? Or a new use environment? Each area where you’re looking to push a boundary is a potential risk to be investigated during feasibility work.
The Model Landscape: A Brief Orientation
Once the key risks have been identified, the next step is to select the right tools -often models- to investigate them. In the context of drug delivery feasibility, models broadly fall into three categories.
In silico models – computational and mathematical approaches – range from simple physics-based calculations through to complex simulations such as computational fluid dynamics (CFD) or finite element analysis (FEA). At their best, they are fast, flexible, and low cost to iterate, making them well suited to early-stage exploration. Their limitation is that they are only as good as the inputs you give them, and at feasibility stage those inputs are often hugely uncertain.

In vitro models – bench-based physical testing – provide empirical data that computational models simply cannot. Synthetic tissue analogues, flow rigs, and bench prototypes all fall into this category. They offer a tangible way to test device and formulation behaviour under controlled conditions and are generally more accessible and affordable than animal or human studies.

Ex vivo models – testing in excised biological tissue – offer real tissue behaviour without the complexity and cost of full animal studies. For early feasibility work involving novel delivery routes or target tissues, ex vivo models are particularly valuable: they can provide rapid, biologically relevant data to characterise tissue properties and validate computational predictions at a stage where in vivo work would be premature.
In practice, the most effective feasibility programmes don’t rely on a single model type. The real skill is in knowing which combination to use, and in what order – and that starts with asking the right questions.
Framing the Right Questions: A Worked Example
Having the right models available is only half the challenge. The bigger risk in early feasibility work is asking the wrong questions of them, reaching for high-fidelity simulation before the problem is properly understood, or pursuing a level of detail that isn’t yet warranted. A multiphysics model of tissue mechanics or a full physiologically-based pharmacokinetic (PBPK) simulation might ultimately be the right tools but deploying them before the fundamental questions have been framed correctly is an expensive way to generate false confidence.
A more effective approach starts not with the model, but with the decision that needs to be made. At each stage of feasibility, it is worth asking: what do we need to demonstrate right now? What is stopping us from moving forward? Once that decision is clear, the next step is to identify the key drivers – what are the dependencies, and do we have reliable data for them? Only then can a model, or set of models, be properly selected, and the guiding principle should always be to choose the simplest approach that can answer the question with the necessary degree of confidence.
To illustrate how this plays out in practice, consider the development of a novel device for delivering drug directly to a target structure within the brain, a scenario that was once thought to sit firmly at the impossible end of the feasibility spectrum. There are many questions that could be addressed during feasibility, but we’ve selected three unrelated questions that have been posed to us in the past as examples of our approach to feasibility:
- What is the delivery force?
- Will the device achieve the required formulation distribution in tissue?
- How will the device influence the clinical effect?
1. What is the delivery force?
The initial instinct may be to model the delivery force in full using a high-fidelity simulation incorporating device mechanics, friction, fluid dynamics, and tissue backpressure simultaneously. However, at this early stage, information about the conditions of delivery e.g. tissue properties, fluid interactions, variability (patient, user, device); may not be fully known. As such, reframing the question reveals a more useful starting point: the real need wasn’t to predict the exact force profile, but to understand whether the force required to deliver the drug would exceed what a typical user could reasonably apply. With that decision in mind, a more pragmatic model strategy emerges; ex vivo tissue characterisation to measure backpressure and understand fluid-tissue interactions, early bench testing with a simple prototype to assess injection force, and a low-order physics-based mathematical model to combine these inputs. Fast to generate, easy to iterate, and sufficient to answer the question that actually mattered at this stage.
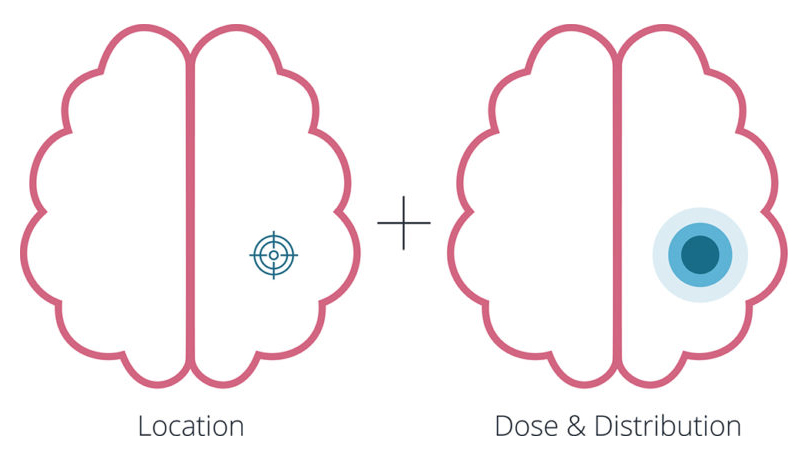
2. Will the device achieve the required formulation distribution in the tissue?
The initial expectation might be a complex computational fluid dynamics (CFD) and fluid-structure interaction (FSI) simulation with a nonlinear, anisotropic tissue model and multiphase flow, an approach that would be computationally expensive and heavily dependent on tissue property inputs that aren’t yet fully known. Reframing shifts the question to something more tractable: can the device achieve the required distribution area (or volume) in tissue? This opens up a staged model strategy; simplified flow and porous media models for rapid screening, followed by testing in brain tissue analogues developed from published academic literature, allowing findings to be benchmarked against existing research. Targeted CFD is then reserved for where refinement is genuinely needed. Lower cost, easier to verify, and designed to work even when input data is uncertain.
3. How will the device influence the clinical effect?
Building a full PBPK model from preclinical data is an understandable ambition, but difficult at feasibility stage, where the biological inputs required are often unavailable or unreliable. Reframing the question from predicting clinical efficacy to “can the device achieve sufficient therapy exposure for target absorption?” enables a more practical, modular approach. Mechanistic models (which describe system behaviour) can be used to characterise device-to-delivery behaviour. Simplified transport models and targeted experiments address delivery-to-distribution as described above. And for distribution-to-exposure, findings can be bridged to existing pharmacokinetic and pharmacodynamic (PK/PD) models (the mathematical frameworks that describe how a drug moves through and acts on the body) making it straightforward to hand off to specialist teams when the time comes. This keeps the focus on the decision at hand and avoids reliance on uncertain biology.
In each case, the pattern is the same: resist the pull towards complexity, reframe the question to be asked around the decisions to be made, and choose the simplest model that can answer it. Models don’t just answer questions, used well, they help reveal the questions worth asking in the first place.
Conclusion
Even the most complex drug delivery challenges can be broken down into manageable pieces. The key is phase-appropriate pragmatism; understanding where your development sits on the feasibility spectrum, selecting models that are fit for the question rather than fit for the complexity, and knowing when you have enough information to make the next decision.
The worked example above illustrates how this plays out in practice. What was once considered an impossible development became feasible through the efficient combination of analytical, synthetic and tissue models, each chosen not for its sophistication, but for its ability to answer a specific, well-framed question at the right stage of the programme. It is an approach we have refined across many such projects, and one that we continue to apply wherever a development pushes beyond the boundaries of what existing platforms and precedent can answer.
To accelerate innovation, we need prompt decision making. Obtaining feasibility answers in weeks rather than years allows redirection of resource toward where it’s most needed (e.g. reframing of therapy or administration route), ensuring effective treatments reach patients faster. The framework works precisely because it is designed to find the answers that matter efficiently.
The tools are available. The challenge, and our experience, is in deploying them wisely.
Connect with CDP
For more on how to accelerate novel and targeted drug delivery feasibility using computational, synthetic, and tissue models to de-risk combination product development, contact Cambridge Design Partnership.
Featured Insights

Precision Delivery: The Missing Link In Cell & Gene Therapy
Drug Delivery to the Brain: Engineering Precision Across Novel Modalities