
Pace is accelerating for tackling neurodegenerative diseases. Can we unlock better treatment? Can we reach a cure?
Ageing populations face neurodegenerative conditions, such as Alzheimer’s Disease, Parkinson’s Disease, Motor Neurone Disease, Multiple Sclerosis, and others. These impact an estimated 60 million people worldwide, equivalent to the current UK population.
Whilst each condition has different mechanisms of neurodegeneration, they all have something in common: prognosis is bleak, treatment is limited, and there is no cure.
However, after decades of research, there has been a series of breakthroughs. Here, we focus on two areas of progress: how treatments have moved on and hope for the future.
The rise of RNA-based therapeutics
The effective development of RNA-based vaccines during the COVID-19 outbreak catapulted RNA-based therapeutics into the spotlight. Whilst theoretical knowledge of RNA therapy has existed for over 30 years, the bulk of associated FDA approval for treatments involving the nervous system has occurred in the last decade(1).
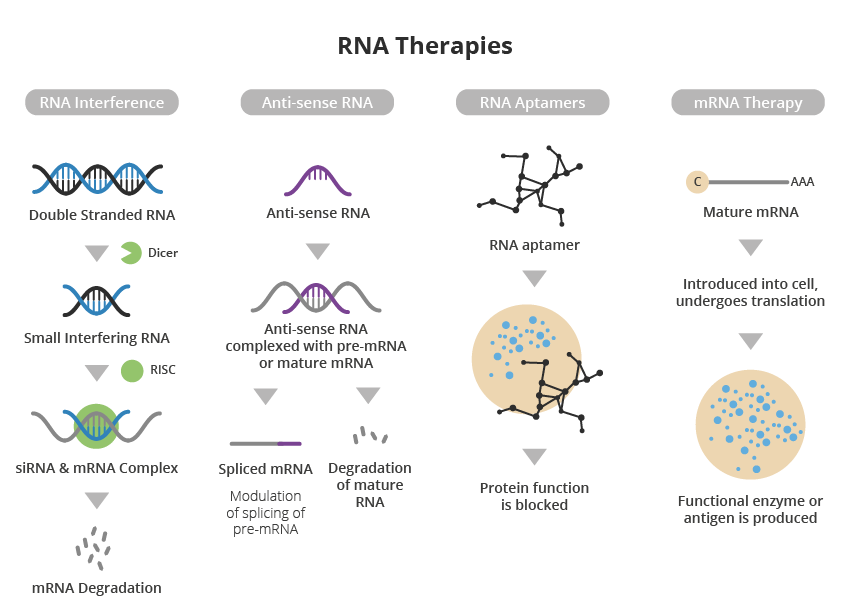
A major advantage of RNA-based therapy over conventional small molecule and protein-based approaches is its high specificity and precision, resulting in a more targeted approach to treating disease with specific gene mutations or overexpression.
However, to devise effective RNA-based therapeutics, the genetic hallmarks of the neurodegenerative disease of interest must be known.
Motor Neurone Disease (MND) is one such condition where specific mutations in the SOD1 gene have been identified and in this case, in two per cent of diagnosed cases.
A recent breakthrough in phase three clinical trials targeted this gene using the drug Tofersen. Tofersen, developed by Biogen, directly interferes with the faulty overproduction of SOD1. After six months, patients had a reduction in SOD1 levels, and after 12 months the same patients reported better mobility and lung function(2,3). Although patients with SOD1 mutations only represent two per cent of those living with MND, these trials provide ‘proof of concept’ that similar gene therapy-based approaches may help other forms of the disease.
Another pioneering strategy, developed by Atalanta Therapeutics and Genentech, focuses on a technology called branched siRNA (small interference RNA). This is a type of molecule that helps regulate gene expression by binding to a complementary messenger RNA, which in turn can encode the gene of interest.
Branched siRNA uses novel RNA interference nucleotide technology to suppress the activity of genes that function abnormally, such as mutations. This slows the progression of the disease or stops it altogether.
It is hoped this approach can be applied across multiple neurodegenerative diseases, including Parkinson’s Disease, Huntington’s Disease and Alzheimer’s Disease.
Although testing is still in the pre-clinical stage, the branched siRNA platform aims to enable RNA interference to be deployed as a therapeutic approach throughout the brain and spinal cord. This overcomes the long-standing challenge of achieving adequate distribution within the central nervous system (CNS) to ensure the therapeutic agent reaches the nervous tissue(4,5).
Progress in non-RNA therapeutics
Non-RNA therapeutics for neurodegenerative conditions also continue to progress. Examples include the monoclonal antibody Donanemab, developed by Eli Lilly. Phase three clinical trials showed it to slow clinical decline by 35% in patients with Alzheimer’s Disease, compared to a placebo(6).
Effective delivery remains a major challenge
One of the main challenges in developing RNA therapeutics, and therapeutics for the brain in general, remains the efficiency of its delivery to the target tissue.
To treat neurodegenerative conditions, the therapeutic agent aims to reach the CNS. The presence of the blood-brain barrier (BBB), a cell-formed wall separating the bloodstream and the CNS, makes it difficult to deliver drugs. The BBB’s almost impermeable characteristics allow very few molecules to cross and make systemic drug delivery less efficacious.
There are two common approaches to overcome this: re-engineering the therapeutic agent to make it compatible with BBB permeability or bypassing the BBB altogether.
Re-engineering the therapeutic agent
This typically involves chemical modification of the drug (e.g., from water-soluble to lipid-soluble molecules) to enable passive diffusion through the BBB. Another approach is to design drug carriers that mimic the structure of endogenous molecules (e.g., monosaccharides, hormones) to activate carrier-mediated transport or nanocarriers(7,8). Both approaches add complexity to manufacturing.
Another cross-BBB approach is Focused Ultrasound (FUS), where high-intensity sound waves temporarily disrupt the BBB to enable drug-loaded microbubbles to enter the CNS9.
Bypassing the blood-brain barrier
Bypassing the BBB can save time and effort in formulation by using a range of therapeutic agents not constricted by size or BBB compatibility. Of its three most common types of delivery: intraparenchymal, intranasal, and cerebrospinal fluid (CSF) delivery; the latter is often the favored approach, due to lower clinical complexity10.
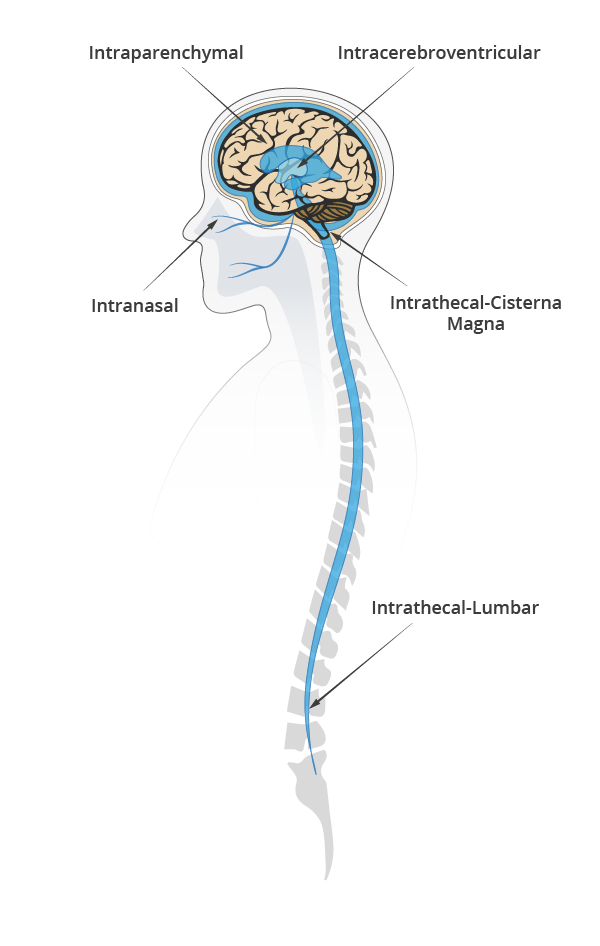
Evaluating CSF delivery routes
CSF delivery most commonly include intrathecal (IT) or intraventricular (ICV) routes.
IT involves an injection either on the lumbar or a cisterna magna region to deliver the drug and let CSF pulsatile flow support the distribution of the therapeutic agent in the brain and spinal cord.
ICV is more invasive. It involves two surgical interventions, one to place a catheter connecting the cerebral ventricles to the injection port at the top of the skull and one to remove the catheter.
To date, ICV has two approved drugs (Rituxan for CNS Lymphoma, and Brineura for Neuronal Ceroid Lipofuscinoses type two). IT lumbar injection has one (Spiranza for Spinal Muscular Atrophy) and plenty more in clinical and pre-clinical stages across a spectrum of neurodegenerative and neurological diseases(11). Irrespective of the approach, the trend is clear: less invasive, lower dosage, and targeted delivery is the way to go.
In the race to show safety and efficacy with either invasive or non-invasive approaches, all solutions will have to be patient-centered.
A new dawn for the treatment of neurodegenerative diseases
The complexities of neurodegeneration have long frustrated scientists and clinicians alike, despite decades dedicated to studying its diseases, aetiologies, and treatments. However, we are making more rapid and more significant progress.
We have some way to go, but we mustn’t overlook the magnitude of these milestones. New therapeutics and delivery techniques are paving the way to more effective and efficient treatment.
By increasing our understanding of genetic hallmarks of the diseases, and using tools such as AI in drug discovery, we can unlock faster pathways to RNA-based treatments. Similarly, by finding innovative ways of demonstrating the safety and efficacy of delivery methods, such as modeling, we can edge closer to less invasive procedures and lower dosages to minimize potential side effects.
We need more research, more awareness, earlier diagnosis, and a better understanding of risk factors to enable prevention and earlier intervention.
But we are now getting closer to better treatment and one day finding a cure.
References
- http://nectar.northampton.ac.uk/16015/1/Anthony_Karen_RNAB_2022_RNA_based_therapeutics_for_neurological_diseases.pdf
- https://www.sheffield.ac.uk/neuroscience-institute/news/promising-mnd-drug-helps-slow-disease-progression-and-benefits-patients-physically
- https://www.nejm.org/doi/full/10.1056/NEJMoa2204705
- https://www.gene.com/stories/pioneering-novel-therapeutics-in-neuroscience
- https://www.nature.com/articles/s41587-019-0205-0
- https://clinicaltrials.gov/ct2/show/NCT04437511?term=TRAILBLAZER-ALZ&cond=Alzheimer+Disease&draw=2&rank=3
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8905930/
- https://ijponline.biomedcentral.com/articles/10.1186/s13052-018-0563-0#:~:text=Modification%20of%20the%20drug%20to,capable%20of%20crossing%20the%20BBB.
- https://clinicaltrials.gov/ct2/show/NCT03321487
- https://www.frontiersin.org/articles/10.3389/fnagi.2019.00373/full
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9305158/
Connect with CDP
For more on how to advance RNA therapeutics and targeted CNS drug delivery for neurodegenerative diseases, contact Cambridge Design Partnership.